par Margaret Helder,
PhD
Creation-Evolution Headlines (traduction Fabrice Bect)
Ce n'est pas un secret parmi les biologistes que les déclarations d'ENCODE II en 2012 étaient controversées. Des scientifiques internationaux du consortium ENCODE ont fait des déclarations sur la façon dont 80% du matériel génétique humain était "fonctionnel"[1] mais le terme fonctionnel, au cours des deux dernières décennies, était devenu associé à des concepts évolutionnistes. L'idée dominante était que les séquences d'ADN dans la cellule n'étaient pour la plupart pas fonctionnelles mais plutôt constituées d'un "ADN poubelle" qui témoignait de l'origine évolutive du matériel génétique de la cellule. Ainsi, l'équipe ENCODE II s'est trouvée très impopulaire auprès de nombreux collègues biologistes pour avoir suggéré un haut niveau de fonctionnalité dans le génome. Maintenant, cependant, en 2020, un troisième projet ENCODE a été publié. S'appuierait-il sur les conclusions de l'équipe ENCODE II, ou réviserait-il ses estimations de fonctionnalité de manière drastique à la baisse ?
Tout a commencé en 2000, lorsque le Président Bill Clinton et le Premier ministre britannique Tony Blair ont annoncé conjointement qu'une première étape du Human Genome Project était terminée. Depuis 1990, une équipe internationale de scientifiques tentait de décrire l'ordre précis des nucléotides dans les chromosomes des cellules humaines. L'idée était qu'un gène fournit l'ordre correct des nucléotides à coder pour une protéine. Les protéines sont des molécules élaborées qui constituent la plupart des composants des cellules vivantes. Des milliards de cellules composent notre corps. On s'attendait à ce que le Human Genome Project fournisse des informations sur environ 100 000 gènes différents.
Imaginez la surprise de tout le monde lorsque les scientifiques ont annoncé qu'il n'y avait qu'environ 23 000 (ou moins) gènes dans les plus de 3 milliards de nucléotides du génome humain. Cela signifiait que moins de 2% du génome humain codait pour des protéines ! Que faisait le reste de cette énorme collection de nucléotides non codants ? Cette découverte semblait indiquer que la grande majorité de l'ADN humain n'avait aucune fonction apparente. Selon Ewan Birney, directeur de l'Institut européen de bioinformatique à Cambridge, en Angleterre, les gens ont toujours su qu'il devait y avoir une certaine régulation des gènes, mais même si les informations utiles étaient doublées, par rapport à la quantité dans les gènes, cela ne ferait que résulter dans 8 à 9% d'informations fonctionnelles au plus parmi les 3,2 milliards de nucléotides.
La vue évolutive des rebuts
indésirables
Il y avait deux réponses
possibles à cette découverte étonnante. L'une consistait
à déclarer que le génome humain était rempli d'"ADN
poubelle". Selon ce point de vue, le génome était
évidemment le produit de longues périodes où l'ADN, qui
avait été à l'origine utile, était maintenant
endommagé, de sorte que d'autres séquences devaient prendre le
contrôle. D'énormes zones de séquences
répétées semblaient confirmer l'idée d'un ADN
inutile. Le génome humain a été déclaré
être une confirmation crue de la réalité de
l'évolution et un rejet de la création. Quel créateur
laisserait traîner autant de déchets ? Et c'est ainsi que l'ADN
poubelle sans fonction a été assimilé à des
arguments en faveur de l'évolution. Un autre groupe a demandé ce
que faisait le reste du génome. Ces longues séquences
étaient-elles fonctionnelles, et si oui, que faisaient-elles
réellement ?
 |
L'évolution darwinienne repose sur des mutations aléatoires comme matière première pour la sélection naturelle. |
L'approche de la recherche
fonctionnelle
Poussé par la curiosité
pour la nature du génome, un consortium international de scientifiques de
10 pays a lancé en 2003 une investigation systématique sur 1% du
génome humain. Tout ce qu'il y avait dans cette étendue de 1%
d'ADN a été étudié, qu'il s'agisse d'un gène
(codant pour une protéine) ou d'une séquence non codante de
nucléotides. Une telle étude devrait également donner une
indication représentative de ce qui se passe dans le reste du
génome. C'était donc en 2007, que la ENCODE (ENCyclopedia of DNA
Elements) a publié ses résultats. À la surprise de
beaucoup, la plus grande partie de l'ADN étudié semblait avoir une
fonction, ou du moins était copiée dans d'autres molécules
de la cellule. Les scientifiques ont ainsi conclu : "la simple vue du
génome comme ayant un ensemble défini de loci isolés
[gènes] transcrits indépendamment ne semble pas
exacte".[2]
Le rapport initial ENCODE était suffisamment intéressant pour encourager le National Human Genome Research Institute basé aux États-Unis à financer une étude sur l'ensemble du génome humain. En conséquence, en 2012, un nouveau consortium ENCODE plus important a publié ses résultats. En résumé, ils ont constaté que :
Les vastes régions désertiques sont désormais peuplées de centaines de milliers de caractéristiques qui contribuent à la régulation des gènes. Et chaque type de cellule utilise différentes combinaisons et permutations de ces caractéristiques pour générer sa biologie unique.[3]
Ce fut une bombe majeure qui devait sortir de l'étude ENCODE II. En fait, le résultat avait été évoqué dans le premier rapport, à savoir une répudiation du concept d'"ADN poubelle". De nombreux biologistes ont été profondément irrités à l'idée que : "Ces données nous ont permis d'assigner des fonctions biochimiques à 80% du génome, en particulier en dehors des régions codant pour les protéines bien étudiées."[4] Un commentaire dans le même numéro renforçait ce point : "Pourquoi l'évolution conserverait-elle de grandes quantités d'ADN "inutile" était restée un mystère, et semblait peu économique. Il s'avère cependant qu'il existe de bonnes raisons de conserver cet ADN."[5]
Un grand nombre d'autres scientifiques dans le domaine des études ADN ont alors déclaré publiquement s'opposer à tous les aspects de l'étude ENCODE. Pour commencer, ils n'aimaient pas la nature systématique de la recherche. Les études systématiques signifient que les chercheurs abordent la recherche sans attentes quant à ce qu'ils vont trouver. En conséquence, toutes les observations sont également les bienvenues puisqu'elles n'ont pas d'idées préconçues. De nombreux scientifiques préfèrent cependant voir des projets fondés sur des hypothèses. Dans ce cas, le chercheur pose une question précise. Les détails non inclus dans la portée de la question, ne seront pas nécessairement respectés. Certains scientifiques éminents ont déclaré que le consortium ENCODE avait commis une erreur en ne posant pas de question basée sur l'évolution.
Parmi les commentaires négatifs sur ENCODE, il y avait Sean Eddy (actuellement à Harvard) qui a dit dans son blog Cryptogenomicon "ENCODE Says What ?"
Personnellement, je ne pense pas que nous puissions comprendre les génomes à moins d'essayer de reconnaître tous les processus évolutionnaires différents, bruyants, et neutres qui y sont à l'oeuvre.[6]
Mais la personne la plus connue pour ses attaques contre ENCODE était Dan Graur. Concernant l'allégation de fonctionnalité à 80% du consortium pour le génome humain, Dan Graur et ses collègues ont déclaré :
Les progrès dans la compréhension de la signification fonctionnelle des séquences d'ADN ne peuvent être atteints qu'en n'ignorant pas les principes évolutionnaires. [7]
Comme alternative, Graur et ses amis ont plaidé pour une proportion fonctionnelle de 10% dans le génome.[8] Leur article s'intitulait de manière provocante : "Sur l'immortalité des téléviseurs : "Des fonctions" dans le génome humain selon l'évangile sans évolution d'ENCODE." [9]
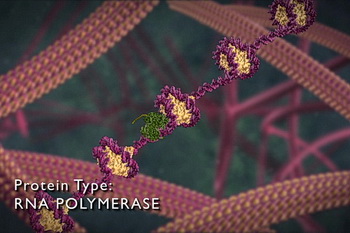 |
ARN polymérase (vert) transcrivant l'ADN (Illustra Media). Pourquoi une cellule travaillerait-elle si dur pour transcrire, éditer, copier et emballer des fichiers indésirables ? |
Contre-arguments et autres
indices
Certains membres du consortium ENCODE
assiégé ont répondu aux attaques dans un article paru dans
les Proceedings of the National Academy of Sciences. Ils ont passé
en revue "les forces et les limites des approches biochimiques,
évolutives et génétiques pour définir des segments
d'ADN fonctionnels, les sources potentielles des différences
observées dans la couverture génomique estimée et les
implications biologiques de ces
écarts."[10] Mais ils ne se distanciaient pas de leurs conclusions antérieures. Ils
ont ainsi évoqué "une activité omniprésente sur une
fraction étonnamment importante du génome, y compris les
régions non codantes et non conservées et les
éléments répétés. Ces résultats
augmentent considérablement les estimations de la limite
supérieure des séquences
fonctionnelles."[11]
Dans sa réponse, cependant, le consortium ENCODE II a évoqué une autre bombe liée à son rapport : le fait que depuis 2005, d'importants marqueurs génétiques liés à des maladies spécifiques, étaient de plus en plus identifiés dans la partie non codante du génome (c'est-à-dire le partie non liée à des gènes, supposément représentant de l'ADN poubelle). Le consortium a utilisé en partie des études d'association à l'échelle du génome (GWAS) pour effectuer ces identifications. Pendant ce temps, des progrès dans la capacité d'obtenir des séquences de génomes entiers de plus en plus de personnes se sont développés. Les spécialistes ont commencé à rechercher des marqueurs uniques chez les personnes atteintes d'une maladie (ou d'un état) spécifique par rapport à d'autres personnes dépourvues de ce trait. Et surprise, surprise :
Plus récemment, des études d'association à l'échelle du génome ont indiqué que la majorité des locus associés aux caractères, y compris ceux qui contribuent aux maladies et à la sensibilité humaines, se trouvent également en dehors des régions codant pour les protéines. Ces résultats suggèrent que les régions non codantes du génome humain abritent un riche éventail d'éléments fonctionnellement significatifs avec diverses fonctions de régulation et autres.[12]
Les scientifiques avaient découvert qu'une activité importante était en cours dans les régions non codantes du génome. Si des marqueurs de la maladie ne sont pas liés à des gènes, cela doit signifier que quelque chose d'important se passe dans ces régions non codantes. Si la zone n'était pas importante, un changement dans l'ordre ADN des nucléotides n'aurait pas d'importance dans un sens ou dans l'autre. Mais cela importait manifestement.
Sources de maladies
génétiques dans l'ADN non codant muté
Lorsque les scientifiques ont
commencé à étudier le génome humain en
détail, on s'attendait à ce que la cause de nombreuses maladies
génétiquement déterminées soit résolue en
étudiant la protéine pertinente et son codage dans un gène.
Cela est certainement vrai pour certaines maladies. Par exemple, l'anémie
falciforme est causée par une mutation dans le 17e nucléotide dans une chaîne de 441 nucléotides codant pour la
chaîne bêta-globine de l'hémoglobine. Cette erreur modifie la
forme de la molécule entière et les résultats peuvent
être catastrophiques pour les personnes qui possèdent la mutation.
De même, l’espoir était que les causes de la plupart ou de
toutes les maladies ou anomalies du développement d’origine
génétique seraient résolues par ces études.
Cependant, les résultats se sont révélés
décourageants jusqu'à ce que les scientifiques commencent à
examiner le GWAS pour de grandes populations. Ces études leur ont permis
de dépasser l'examen de gènes spécifiques afin de
rechercher où les mutations pertinentes se produisaient réellement
dans la molécule d'ADN.
ENCODE III apporte plus de
lumière
Ces deux bombes d'une forte
proportion de fonctionnalité dans tout le génome et la
localisation de la plupart des marqueurs associés à la maladie
dans les parties non codantes du génome, sont des thèmes majeurs
vigoureusement promus dans ENCODE III, huit ans après ENCODE
II.[13] Sur la
question des marqueurs de maladies, un article d'ENCODE III déclare que
"les études de GWAS et de génomique du cancer continuent de
déposer des variations de séquence liées à la
maladie dans des bases de données publiques, et la plupart de ces
variantes tombent dans des régions non
codantes.[14] Réfléchissant à ce fait, une équipe du consortium
souligne que nous percevons désormais "une caractéristique
fondamentale de l'architecture génétique de la maladie qui a
jusqu'ici, à notre connaissance, échappé à
l'attention".[15] Ce que nous observons maintenant, disent-ils, "a d'importantes implications
théoriques et pratiques pour comprendre à la fois l'architecture
génétique de la maladie et le problème de la connexion des
signaux génétiques avec leurs gènes cibles, ce qui est
essentiel pour la traduction
thérapeutique".[16]
Ces informations démontrent qu'il reste encore beaucoup à apprendre sur les causes génétiques de nombreuses maladies. Apparemment, la majorité des mutations liées à la maladie se situent loin des gènes codant pour les protéines. En conséquence, un aperçu en ligne de l'Encyclopédie ENCODE version 3 déclare : "plus de 80% des variants signalés par le GWAS se trouvent dans des régions non codantes du génome et le mécanisme de leur contribution à l'apparition de la maladie est inconnu."
Implications pour la
fonction
Bien que ces études
liées à la maladie nous pointent vers les sections
régulatrices (non codantes) du génome, la question vraiment
critique est de savoir dans quelle mesure le génome est fonctionnel. Pour
commencer, il serait utile que tout le monde s'entende sur ce que signifie le
mot "fonctionnel". Les gens d'ENCODE II ont supposé que toute
séquence d'ADN copiée dans une autre molécule devait
être fonctionnelle. Dan Graur s'est opposé à cette
hypothèse, déclarant que "la transcription n'équivaut pas
à une
fonction".[17] Son argument était que la simple existence d'une activité
moléculaire n'implique pas nécessairement que l'activité
profite à la cellule ou contribue à son succès
évolutif. Le produit moléculaire peut avoir été
produit accidentellement. L'équipe ENCODE III n'a pas reculé par
rapport aux positions précédentes du consortium. Ils ont
continué à défendre l'idée que toute séquence
nucléotidique menant à un produit moléculaire ou à
toute activité chimique est
fonctionnelle.[18]
Alors que l'équipe ENCODE II a commis l'erreur tactique de citer une valeur élevée pour la fonctionnalité du génome, la nouvelle équipe n'a pas commis une telle erreur. À bien des égards, cependant, ils ont indiqué qu'ils étendaient en fait les conclusions d'ENCODE II. Ils ont déclaré : "Il est important de noter que bien que de très nombreux éléments non codants aient été définis, l'annotation fonctionnelle des éléments identifiés par ENCODE n'en est qu'à ses débuts."[19] Il y aura d'autres révélations de ce genre à venir. En outre, ils insistent : "nous ne prétendons pas que le schéma de classification actuel des cCRE [régions de régulation] reflète le spectre biologique complet des activités de régulation codées dans le génome."[20]

Le noyau d'une cellule humaine
contient au moins 6 pieds d'ADN.
Cliquez pour voir Illustra Media, "18 billions
de pieds à vous".
Perspectives
d'avenir
Il y aura sans aucun doute d'autres
découvertes à venir. ENCODE III a ajouté à la preuve
de la fonction dans l'ADN non codant avec des séquences de
régulation nouvellement découvertes telles qu'une "classe
d'éléments de séquence fonctionnelle non reconnus
auparavant par
ENCODE".[21] Le
résultat de cette seule découverte est : "Ces données
élargissent le catalogue d'éléments fonctionnels
codés dans le génome humain par l'ajout d'un grand ensemble
d'éléments qui fonctionnent au niveau de l'ARN en interagissant
avec les protéines de liaison à
l'ARN."[22] De
même, ils déclarent : "les données d'ENCODE III ont
augmenté le nombre d'éléments cis-régulateurs de
près de 22% par rapport à ENCODE
II."[23] Ils
ont fait des déclarations similaires tout au long du rapport, par exemple
sous le titre "Encodage dense d'informations
régulatrices".24
De toute évidence, l'équipe ENCODE III étendait l'identification des éléments fonctionnels beaucoup plus loin que ne l'avait fait ENCODE II. Leur déclaration récapitulative confirme la conviction que les arguments en faveur de la fonction génomique dans l'ADN non codant se renforceront :
Il est devenu évident que, par pratiquement n'importe quelle métrique, les éléments qui régissent la transcription, l'organisation de la chromatine, l'épissage et d'autres aspects clés du contrôle et de la fonction du génome sont codés de manière dense dans de nombreuses parties de la séquence du génome humain.[24]
Leçons
apprises
Les équipes d'ENCODE III n'ont
pas reculé face à la pression des évolutionnistes
doctrinaires. Elles ont continué à faire leurs observations et
à laisser les preuves parler d'elles-mêmes. En
général, elles ont adopté une approche pragmatique de
l'ensemble de la question dans l'espoir que "Collectivement, les données
et le registre ENCODE fournissent une ressource considérable pour la
communauté scientifique afin de mieux comprendre l'organisation et la
fonction des génomes humains et
murins."[25] Ils ne ressentaient aucune obligation de présenter des théories
évolution nistes non pertinentes sur l'ADN indésirable, d'autant
plus que celles-ci ont déjà été
réfutées par les résultats d'ENCODE.
nistes non pertinentes sur l'ADN indésirable, d'autant
plus que celles-ci ont déjà été
réfutées par les résultats d'ENCODE.
Margaret Helder a terminé ses études avec un doctorat en botanique de l'Université Western à London, Ontario (Canada). Elle a été embauchée comme professeure adjointe en biosciences à l'Université Brock à St. Catharines, en Ontario. Arrivé en Alberta en 1977, le Dr Helder était un témoin expert pour l'État de l'Arkansas, en décembre 1981, lors du procès de "traitement équilibré" du débat création/évolution. Elle a été membre du comité de rédaction de Occasional Papers du Baraminology Study Group en 2001. Elle a également donné des conférences une ou deux fois par an (sur invitation) dans des cours réguliers à l'Université de l'Alberta (St. Joseph's College) de 1998 à 2012. Ses publications techniques comprennent des articles dans le Canadian Journal of Botany, chapitre 19 de Recent Advances in Aquatic Mycology (EB Gareth Jones. Editor. 1976), et plus récemment, elle a écrit No Christian Silence on Science (2016) qui promeut l'évaluation critique des allégations scientifiques. Elle est mariée à John Helder et ils ont six enfants adultes.
Notes
[1] - ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489 #7414: 57-74.
[2] - ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 #7146: pp. 799- 816. See p. 812.
[3] - Brendan Maher. 2012. The Human Encyclopaedia. Nature 489 #7414 pp. 46-48. Voir p.
[4] - ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489 p. 57.
[5] - Ines Barroso. 2012. Non-coding but functional. Nature 489 p. 54.
[6] - 8 septembre, 2012.
[7] - Dan Graur et al. Genome Biol. Evol. 5 (3): p. 587 italiques ajoutées par moi. Graur et al. 578.
[8] - Genome Biol. Evol. 5 (3): 578-590.
[9] - Manolis Kellis et al. Defining functional DNA elements in the human genome. PNAS 111 #17 pp. 6131-6138. Voir p. 6131.
[10] - Kellis et al. 6134.
[11] - Kellis et al. 6131 italiques ajoutées par moi.
[12] - ENCODE Project Consortium. 2020. Perspectives on ENCODE. 583 #7818: 693-698. And flagship article: Expanded encyclopaedias of DNA elements in the human and mouse genomes. pp. 699-710.
[13] - Fabian Grubert et al. Landscape of cohesin-mediated chromatin loops in the human genome. Nature 583 #7818: PP. 737-743. Voir p. 743.
[14] - Wouter Meuleman et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature 584 #7820: pp. 244-251. Voir 250.
[15] - Meuleman et al. 251.
[16] - Graur et al. 581.
[17] - La séquence de nucléotides "qui spécifie des produits moléculaires (par exemple, des gènes codant pour des protéines ou des ARN non codants) ou des activités biochimiques avec des rôles mécanistes dans la régulation du gène ou du génome (par exemple, des promoteurs ou des amplificateurs de transcription)" est fonctionnelle. p. 699 et il y avait une définition similaire avec ENCODE II p. 57.
[18] - ENCODE Project Consortium. 2020. Perspectives on ENCODE p. 697.
[19] - ENCODE Project Consortium. 2020. Flagship article p. 706.
[20] - ENCODE Project Consortium. 2020. Flagship article p. 702.
[21] - Eric L. Van Nostrand et al. A large-scale binding and functional map of human RNA-binding proteins. Nature. 583 #7818: pp. 711-719. Voir p. 711.
[22] - ENCODE Project Consortium. 2020. Flagship article p. 706.
[23] - Meuleman et al. 247.
[24] - ENCODE Project Consortium. 2020. Flagship article p. 709.
[25] - ENCODE Project Consortium. 2020. Flagship article p. 699.